

Abstract
At the top of the hematopoietic hierarchy are hematopoietic stem cells (HSCs), which reside in the bone marrow and are characterized by the ability to self-renew or differentiate into various types of mature blood cells. The self-renewal capacity of HSCs relies on the accurate transmission of epigenetic marks to their progeny. Our lab has shown previously that, despite global hypomethylation, DNA hypermethylation frequently occurs on Polycomb group protein (PcG) target genes and many tumor suppressor genes in aged HSCs (Sun et al.Cell Stem Cell. 2014). At the same time, such epigenetic marks are correctly maintained in young HSCs. These observations indicate the presence of epigenetic maintenance systems that deteriorate with age. Currently, the molecular mechanisms through which aberrant DNA hypermethylation accumulates only with age are unclear.
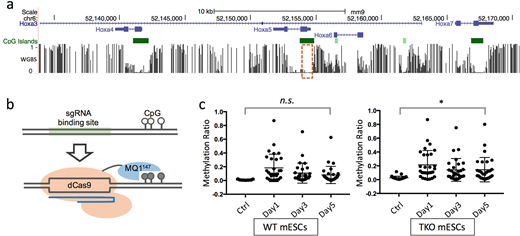
To address this gap in knowledge, we examined the maintenance and clearance of forced DNA methylation in murine embryonic stem cells (ESCs) using a CRISPR/dCas9-based targeted DNA methylation tool, dCas9-MQ1147 (Lei et al, Nature Communication. 2017). We forcibly methylated genes that are bound in ESCs by PcG, including Runx1, Gata2, and Hoxa loci. Surprisingly, we found that the exogenous methylation induced by dCas9-MQ1147 was efficiently removed from the target sites during cell proliferation, indicating that methylation status is predetermined and maintained by local genetic and epigenetic marks.
To understand which demethylation mechanism is responsible for the removal of the exogenous aberrant DNA methylation in our model, we treated both human somatic cells and murine ESCs with cell cycle arrest drugs, including selective ATP-competitive inhibitor of CDK1 (Ro-3306), selective Ca2+/calmodulin-dependent protein kinase inhibitor (KN93), and microtubule formation inhibitor (Paclitaxel) after inducing targeted methylation. Following treatment, we observed that cell cycle arrest cannot delay passive demethylation at the hypermethylated sites, indicating the removel of aberrant methylation is DNA duplication independent. To further investigate the demethylation mechanism herein, we next validated the contribution of DNA hydroxymethylation enzyme Ten-eleven Translocation (TET) activity in the Tet1/2/3 triple knockout (TKO) mESCs. Time-dependent experiments showed that TKO mESCs had a significant exogenous methylation retention compared to their wild-type counterparts. These data indicate that TET family proteins are recruited to remove aberrant methylation from the unmethylated PcG binding region via TDG or base excision repair, but not inaccurate maintenance by DNA methyltransferase 1 (DNMT1).
To determine which TET protein or proteins contribute to the maintenance of predetermined unmethylation status, we used prokaryote DNA methyltransferase MQ1 wild-type protein to generate genome-wide hypermethylation in wild-type mESCs. We detected that TET1 was the most highly upregulated TET protein, with over 5-fold upregulation, following induced hypermethylation. Furthermore, by applying the novel Degron targeted degradation technique, we specific remove the expressed MQ1 protein and found that the degradation of MQ1 led to the reduction of Tet1 overexpression. These data indicate that TET1 participates in the removal of aberrant DNA methylation in mESCs.
Overall, this study suggests that a proofreading mechanism at the PcG-targeted region recognizes aberrant DNA methylation and recruits TET1 to restore its original unmethylated status. The dysregulation of this mechanism in aging HSCs may lead to the accumulation of methylation abnormalities during proliferation.
This study sheds light on an important molecular mechanisms responsible for maintaining the epigenetic status in ESCs and provides insight into how aberrant DNA methylation accumulates in these cells over time.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal